Published on

COVID-19 analysis looks at variant spread
By Davis Suppes | Bond LSC
Variants of the virus that causes COVID-19 continue to plague the world with spikes in infection, keeping the current pandemic from being fully controlled as Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) infections remain unmanageable in some parts of the world.
Researchers at Bond Life Sciences Center and the University of Nebraska Medical Center (UNMC) have been conducting research on mutations in four new variants, B.1.1.7, B.1.351, P.1 and CAL.20C. These evolving variants appear to be more infectious and transmissible than the original Wuhan-Hu-1 virus.
It all started with a new spike in infections of a variant of SARS-CoV-2 in the UK. Despite a slowdown in infections due to precautions of masks and lockdowns, the new spike meant something about the virus had changed.
“Our goal was to find out where these variants are distributed, and how they are migrating,” said Kamal Singh, Assistant Director Molecular Interactions Core, and scientist at Bond LSC. Singh was a corresponding author on the paper and has been at Bond LSC for 12 years.
Their work showed completely different mutations in these variants based on regions, but also where variants have common lineage. That commonality can be seen between the CAL.20C and B.1.351 variants.
While related to each other, they also developed different mutations.
“B.1.1.7 was more infectious and was able to spread to more people. It slowly dominated and that’s what happened in the United States as B.1.1.7 is now the dominant variant of SARS-CoV-2 in the US,” said Saathvik Kannan, a co-author. Kannan is going to be a 10th grader at Columbia’s Hickman High School this fall but works under Singh at Bond LSC as a computer programmer and researcher.
Once they identified the related COVID-19 variants, they wanted to trace their migration. This allows them to see where the mutations are being carried and which regions have a more dense number of infections. The prevalence of variants was key in identifying where they were moving.
Their analysis looked at certain mutations in each variant to see how they were related, why some sequences have mutations that others don’t and tried to determine how they evolved from their original sequence.
By comparing the structure of each virus, they were able to illustrate how the related mutations moved state by state across the country.
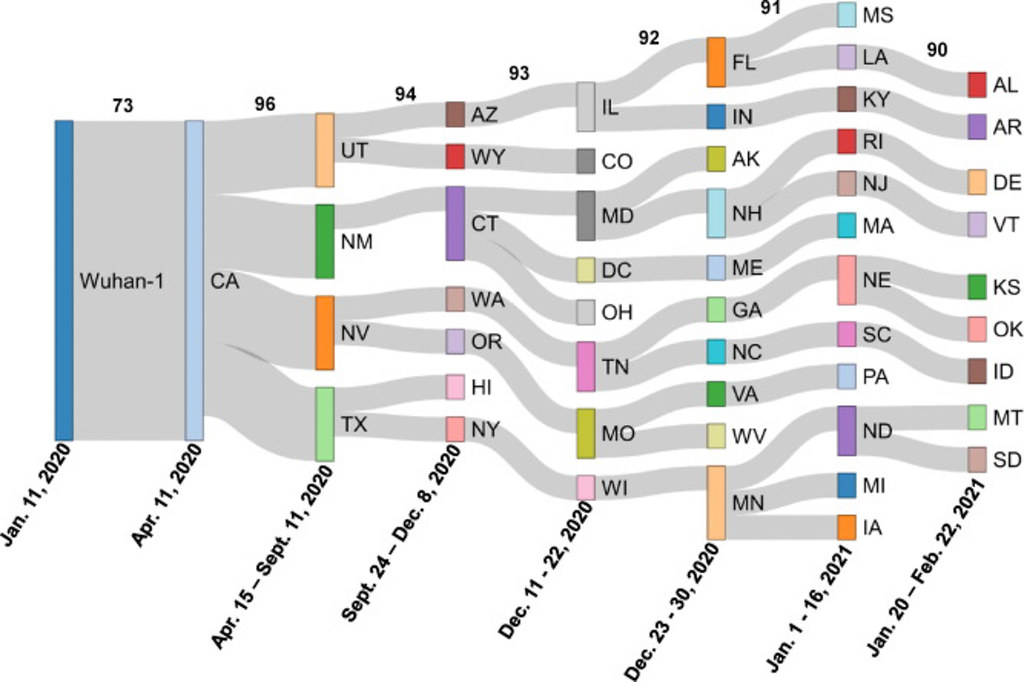
“It is pretty clear that some states have a higher homology match, which helps us see a pretty linear trend of migration from the Western states to the East in the United States,” said Austin Spratt, a computer science senior and lead author on the paper. Spratt has worked under Singh at Bond LSC for two years.
The study cautions that the sequences of viruses are being deposited at an unprecedented rate, so it is possible that some conclusions may change as more sequences of variants become available.
“These variants are continuously evolving and complex in nature, but a proper understanding of what makes each variant unique, its genetic makeup and its interaction with host proteins is critical to developing the most effective vaccine,” said co-corresponding author Siddappa Byrareddy, Professor & Vice-Chair of Research, in the Department of Pharmacology & Experimental Neuroscience, at the University of Nebraska Medical Center. “Not only will this help us to design better vaccines now, but greatly help us to be prepared to deal with more mutant variants in the future.”
Although analysis indicates that mutations only in one variant do not reflect evolution of it, these genetic mistakes may increase infectiousness of a particular variant
The short period of time, volume of variants and limited travel around the world all point to a greater infectivity than the original (Wuhan-Hu-1) virus. Recent deadly surges in India caused by variant B.1.617.2 or the Delta variant will soon be added to their analysis to further understand the COVID-19 pandemic.
Their research was published in the paper ‘Evolution, Correlation, Structural Impact and Dynamics of Emerging SARS-CoV-2 Variants’ in the “Computational and Structural Biotechnology Journal” on June 19, 2021.
Funding provided by: Bond LSC, Swedish research Council, American Lung Association, National Institute of Dental and Craniofacial Research in collaboration with Prof. Gary Weisman of the Bond Life Sciences center.